Massive Human CRISPR-Based Genotype-Phenotype Map Is Now Complete
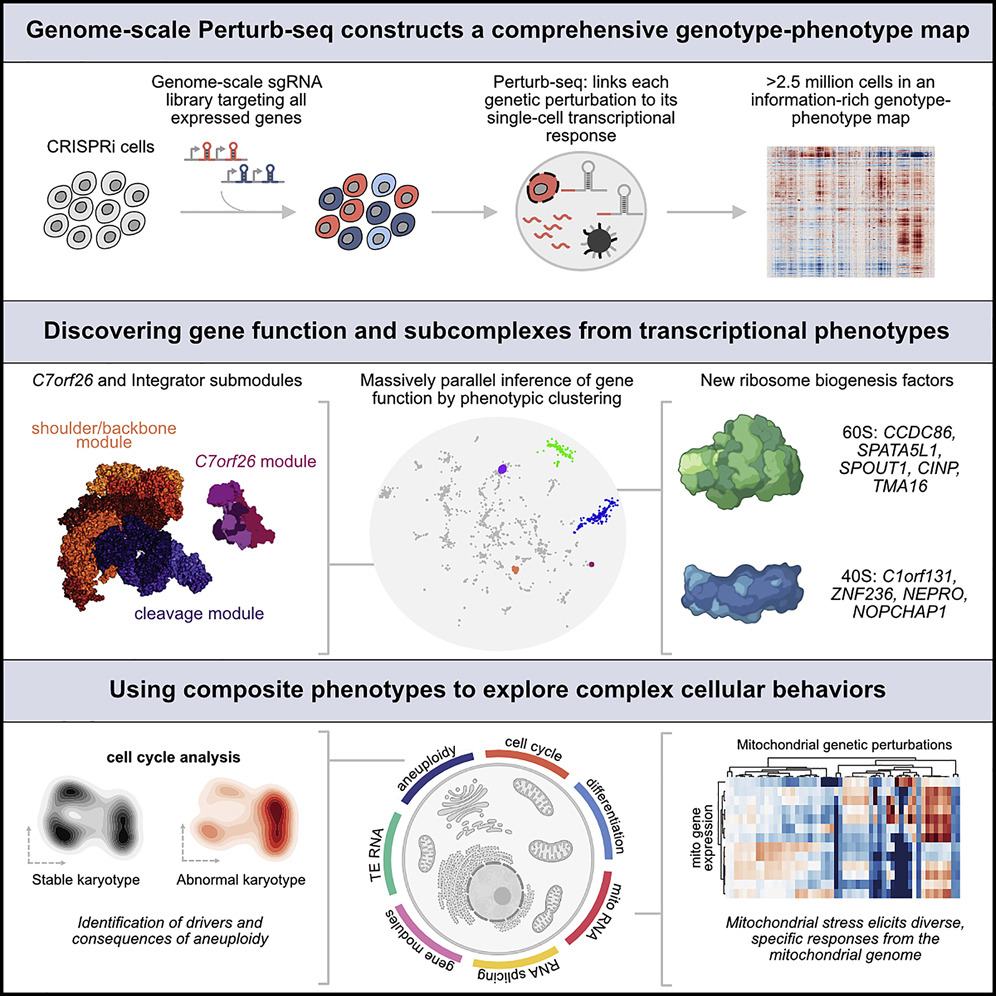
Two decades after the Human Genome Project, scientists have gone beyond mere genetic sequences to present the first comprehensive functional map of genes that are expressed in human cells. New research published in Cell presented results of the genotype-phenotype landscapes with genome-scale Perturb-seq, an experimental approach to map the transcriptional effects of genetic perturbations by combining CRISPR-based genetic screening with single-cell RNA-sequencing.
With the database, researchers can screen for genotype-phenotype relationships without having to perform any experiments. The researchers then used the database to look at cellular effects of genes with unknown functions, investigate mitochondrial stress response, and screen for genes that cause loss/gain of chromosomes, a phenotype that has proved difficult to study in the past.
Related article: Finding New Antibiotics from Bacterial Genes With Computational Biology and Genome Sequencing to Synthesize Novel Drug Molecules
Perturb-seq, Sequencing Cell With CRISPR-Cas9 Genome Editing
The Perturb-seq method, first published in 2016, uses CRISPR-Cas9 genome editing to introduce genetic variations into cells and then utilizes single-cell RNA sequencing to screen information of expressed RNAs resulting from a given genetic change. Because RNAs control every aspect of cell behaviors, the method can decode the many cellular effects caused by genetic changes.
Since the initial 2016’s publication, multiple efforts have been made to scale up the sequencing technology and deploy it in small-scale investigations. In the new study, collaborators expanded the method to the entire human genome. Using human blood cancer cell lines as well as non-cancerous retinal cells, the researchers performed Perturb-seq across over 2.5 million cells and used the data to build a comprehensive map linking genotypes to phenotypes.
Related article: In Vivo Functional Screen, PerturbSeq Helps Understand the Complex Interplay of Autism Risk Genes
Putting the Genotype-Phenotype Map to Use
The authors looked into genes with unknown functions upon completion of the database, because the database also presents phenotypes of many already known genes, they could use the data to compare the data to unknown genes and look for similar transcriptional results, which could piece together the gene products that worked together as part of a larger complex.
Mutation of the C7orf26 gene attracted their attention, they noticed that the gene’s removal led to a similar phenotype that was part of the Integrator protein complex that played a role in creating small nuclear RNAs. The Integrator complex is made up of many smaller subunits – previous studies had suggested 14 individual proteins – and the researchers were able to confirm that C7orf26 made up a 15th component of the complex, demonstrating the power of this new top-down perspective.
“The advantage of Perturb-seq is it lets you get a big dataset in an unbiased way,” says former MIT Weissman Lab postdoc Tom Norman, a co-senior author of the paper. “No one knows entirely what the limits are of what you can get out of that kind of dataset. Now, the question is, what do you actually do with it?”
The researchers hope to use Perturb-seq on cells other than the original cancer cell line in the future, and also hope to continue to explore their map of gene functions. “This really is the culmination of many years of work by the authors and other collaborators, and I’m really pleased to see it continue to succeed and expand,” says Norman.
©www.geneonline.com All rights reserved. Collaborate with us: service@geneonlineasia.com
Author
LATEST
Oncology’s New Drugs on the Horizon (II): Radiotherapy, Radioligands & Other Therapeutic Agents
2024-04-12
Vertex Pharmaceuticals Makes $4.9 Billion Bet on Kidney Disease Treatment through Alpine Immune Acquisition
2024-04-11
Bayer Signs New Partnership with Google Cloud, Joining Hands for AI Solutions for Radiologists
2024-04-10

EVENT
2024-04-20
16th SABPA OC/LA Annual Biomedical Forum
The Beckman Center, 100 Academy, Irvine, CA, 92617